What to do about geneticists? On one hand, they are so smart that we should accept whatever they say, no matter how absurd, inaccurate, or even racist it may be. (See Nicholas Wade’s Before the Dawn).[1] On the other hand, they’re ignorant and arrogant assholes and they should be thrown in jail (See Trofim Lysenko).[2] There has got to be a middle ground.
The gorilla genome is now out, and when combined with human, chimpanzee, and orangutan, it allows us to do a phylogenetic comparison.[3] We have known since the 1980s that human-chimp-gorilla genetically is a very close call, with DNA tending to place humans and chimps a little closer, but only with a lot of discordance or statistical noise. (That is in fact exactly what the ill-fated DNA hybridization showed, although it was infamously misrepresented.) When the mtDNA data first came out [4] they linked human to chimp pairwise, but only if you ignored the fact that over half of the phylogenetically informative DNA sites did not in fact show it to be human-chimp. Those data showed it to be chimp-gorilla and human-gorilla. The only way to extract human-chimp from those data was to treat the question like a Republican primary, where whoever gets the plurality of the votes wins the state. So human-chimp was Mitt Romney, winning the nomination, but with barely 45% of the phylogenetically informative sites.
It then becomes a trivial task to explain away the discordant data, that is to say, the 55% of your data that you have decided is giving you the “wrong” answers. You say it is “incomplete lineage sorting” or the result of ancestral polymorphisms, which have segregated into descendant taxa in a pattern different from the sequence of speciation. Geneticists illustrate this with images that always seem to remind me of maps of the London Underground, with chimpanzees being Bakerloo and humans Victoria Station.
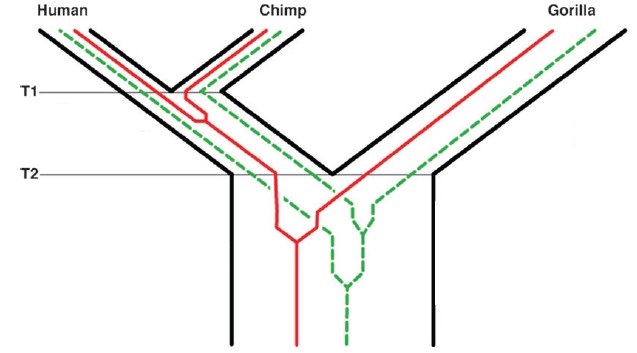
But I digress. It might also be parallel mutation or even backcrossing. The problem, though, is that you have a lot of homoplasy, and one of the assumptions of cladistic/phylogenetic analysis is that homoplasy (i.e., observed as discordance) is very, very low compared to synapomorphy (i.e., the shared derived characters that you think are tracking the actual branching history of the species).
This is the equivalent of simply choosing the most parsimonious solution to the phylogenetic problem. Most of the data that give a pairwise resolution give this pairwise resolution, therefore it must be the right one. But there is an inherent contradiction in this logic. You are choosing the most parsimonious solution in a system that is not obviously very parsimonious. In other words, if you are willing to accept the possibility that 55% of your phylogenetically informative sites are homoplasies (that is to say, are giving you the “wrong” answer), then how can you reject the idea that 70% of your sites might be giving you the “wrong” answer? I talked about this many years ago in the American Journal of Physical Anthropology.[5]
The model that fits the data best is not a model of two successive bifurcations, but what we called at the time a “trichotomy” and now would call “reticulate” or even “rhizotic” evolution.[6] [7]
The geneticists working on this problem have been hampered by the cladistic necessity of regarding speciation as events, rather than as processes – when their ape data are showing speciation as processes, not as events. The new paper on the gorilla genome says that 30% of their phylogenetically informative sites are discordant. This is how the new paper imagines the genomic relationships of humans, chimps, and gorillas – as indicating two temporally isolated speciation “events” and whatever the hell is going on in the middle there.

The creationists jumped all over this inconsistency, and it really is just the result of sloppy thinking by the scientists.
In trying to plug the genomic data into sequential speciation events, we are committing the square-peg-round-hole fallacy. There are historical and ideological reasons for depicting it as two successive, temporally distinct “events,” but that certainly misrepresents the evidence, and most likely misrepresents the biological history. One of the most bizarre illustrations was in a recent introductory textbook, which showed this to students:

It’s trying to say that there were two speciation events, 7 mya and 6 mya, but has located the 7 mya event incorrectly. If you look at the scale, you’ll see that it’s actually drawn at 8 million, to put a separation between them that shouldn’t be there. The same text draws it this way a bit later. with very little (vertical) time separating the two “events” at 7-8 mya and 5-7 mya, but a lot of (horizontal) space. That ought to learn ‘em!

Obviously, that’s not the text I use.
The new paper on the gorilla genome, I might add, sets the “speciation events” at 6.0 and 3.7 mya. The 3.7 mya date for the divergence of human and chimpanzee is simply, to the extent that anything can be falsified in the fossil record, false - although it is oddly congruent with some of Vince Sarich and Allan Wilson’s early writings on the subject in the 1960s.[8] The (myriad) authors of the new paper go on to argue that they can juggle some of the parameters in their computer program to make the dates come out to about 6 and 10 million years ago – as if that is supposed to give us confidence!
For the Alternative Introduction, I drew this figure to illustrate the problem.

Rather than prurient talk about cross-species buggery on the part of early hominids, how about speciation here as a temporal process, and populations through time as anastemosing capillary systems (Earnest Hooton’s metaphor, expressing the same point as rhizomatic and reticulate evolution). It is also noteworthy that we tend to model and depict the gene pools of all three species as equivalent, when we’ve known for years that chimps and gorillas, even as relict populations, have gene pools that are considerably more extensive than that of our own species. That is to say, Homo sapiens is relatively depauperate in genetic diversity. The only study to try and incorporate that information into a phylogenetic analysis, many years ago, found that it completely obscured the phylogenetic “signal” and that it was therefore a fool’s errand to try and extract two successive bifurcations from a genomic analysis of human, chimpanzee, and gorilla.[9]
Interestingly, the new paper actually did look at diversity in gorilla genomes, but didn’t incorporate that into their phylogenetic analysis. Bottom line: Human evolution is probably more interesting than the geneticists realize.
[1] Wade N. 2006. Before the Dawn: Recovering the Lost History of Our Ancestors. New York: Penguin.
[2] Medvedev Z, and Lerner I. 1969. The Rise and Fall of TD Lysenko. New York: Columbia University Press.
[3] Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T et al. . 2012. Insights into hominid evolution from the gorilla genome sequence. Nature 483(7388):169-175.
[4] Horai S, Satta Y, Hayasaka K, Kondo R, Inoue T, Ishida T, Hayashi S, and Takahata N. 1992. Man's place in hominoidea revealed by mitochondrial DNA genealogy. Journal of Molecular Evolution 35(1):32-43.
[5] Marks J. 1994. Blood will tell (won't it?)? A century of molecular discourse in anthropological systematics. American Journal of Physical Anthropology 94:59-79.
[6] Marks J. 1995. Learning to live with a trichotomy. American Journal of Physical Anthropology 98:211-213.
[7] Arnold M. 2009. Reticulate Evolution and Humans: Origins and Ecology. New York: Oxford University Press.
[8] Sarich VM. 1968. The origin of the hominids: An immunological approach. In: Washburn SL, and Jay PC, editors. Perspectives on Human Evolution I. New York: Holt, Rinehart, and Winston. p 94-121.
[9] Ruano G, Rogers, Jeffrey A., Ferguson-Smith, Anne C., Kidd, Kenneth K. 1992. DNA sequence polymorphism within hominoid species exceeds the number of phylogenetically informative characters for a HOX2 locus Molecular Biology and Evolution 9(4):575-586.